Ollier disease
Dr. KS Dhillon
Introduction
Enchondromas are common intraosseous, usually benign cartilaginous tumors. They develop close to the growth plate cartilage. When multiple enchondromas are present, the condition is called enchondromatosis. Enchondromatosis is also known as Ollier disease. The prevalence of Ollier disease is estimated to be 1 in 100,000 individuals. Clinical manifestations usually appear in the first decade of life. In Ollier disease, there is an asymmetric distribution of cartilage lesions. These lesions can be extremely variable in terms of size, number, location, age of onset, and requirement for surgery. Enchondromas can produce skeletal deformities and limb-length discrepancies. They have the potential risk for malignant change to chondrosarcoma. When multiple enchondromatosis is associated with soft tissue hemangiomas, the condition is known as Maffucci syndrome. Ollier disease and Maffucci syndrome occur in isolated patients. These conditions are not familial. It is uncertain whether the disorder is caused by a single gene defect or by combinations of germ-line and/or somatic mutations. The diagnosis is made by clinical and conventional radiological evaluations. Histological analysis has a limited role. It is mainly used if malignancy is suspected. Medical treatment has no role in the management of enchondromatosis. Surgery is usually indicated when there are complications such as pathological fractures, growth defects, and malignant transformation. It is difficult to assess the prognosis for Ollier disease. Forms with an early onset appear more severe. There is a risk of malignant transformation of enchondromas into chondrosarcomas in patients with Ollier disease.
Definition
Enchondromas are common benign cartilage tumors. They develop in the metaphyses and may become incorporated into the diaphyses of long tubular bones, in close proximity to growth plate cartilage (1-3). They are usually asymptomatic. Enchondromatosis or Ollier disease is defined by the presence of multiple enchondromas with an asymmetric distribution of cartilage lesions that can be very variable in terms of number, size, location, and age of onset (4).
Multiple enchondromatosis associated with soft tissue hemangiomas is known as Maffucci syndrome.
Epidemiology
It is the 2nd most common benign cartilage lesion. Osteochondroma is the most common. The male-to-female ratio is 1:1. It is most common in 20-50 year old individuals. It is usually found in the medullary cavity of the diaphysis or the metaphysis. The most common location is the hand (60%). It is more common in the hand as compared to the feet. The most common primary bone tumor in the hand is the enchondroma. Other locations include the distal femur (20%), proximal humerus (10%), and the tibia.
Clinical presentation
Ollier disease usually presents in the first decade of life. Clinical presentation usually starts with the appearance of palpable bony masses on a finger or toe, asymmetric shortening of an extremity, limp, osseous deformities with or without pathologic fractures (1-3). Physical examination usually shows visible masses embedded within phalanges, metacarpal, and metatarsal bones. Enchondromas usually affect long tubular bones, particularly the femur, the tibia, and/or the fibula. Flat bones, especially the pelvis, can also be affected. Multiple bones can be affected and the lesions are usually asymmetrically distributed, predominantly affecting one side of the body. Affected bones are quite often deformed and shortened. Bone shortening may be the only clinical sign of the disease. Bone shortening is often associated with bone bending and curving. This can lead to limitations in joint movements. Forearm deformities that are frequently encountered are similar to those observed in hereditary multiple exostosis (HME). The trunk is usually not affected, except for rib enchondromas and scoliosis which results from pelvis imbalance. In children, the lesions are subjected to pathologic fractures.
Clinical forms
Enchondromatosis has been recognized for a long time. Ollier at the end of the 19th century emphasized the asymmetrical and random distribution of enchondromas. Some authors have distinguished two subtypes of enchondromatosis i.e. enchondromatosis and Ollier disease. The first subtype affects mostly men. In this type, enchondromas are located mainly at the extremities and appear to be transmitted in an autosomal dominant fashion (5). The second form mainly affects women. In this subtype, there is sporadic unilateral distribution of enchondromas. The basis for this classification into two forms is not supported by a thorough analysis of available data. When multiple enchondromas are associated with hemangiomas it is referred to as Maffucci syndrome. Gabos and Bowen recently reported a previously unreported form in which there is extensive involvement of the epiphyseal and metaphyseal regions of long bones of the lower extremity (6).
Radiography
Enchondromas are most likely present at birth although they are rarely observed at birth.
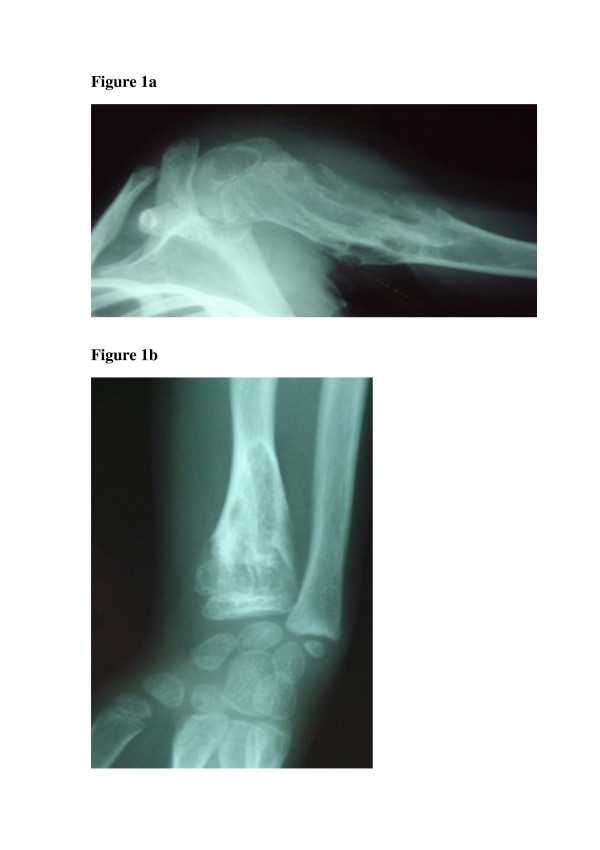
X-rays usually show multiple, radiolucent, homogenous lesions with an oval or elongated shape and a slightly thickened bony margin (fig 1a and 1b) (1-3). The lesions run parallel to the long bone axis. The lesions usually calcify with time. They become diffusely punctated or stippled, and a light trabeculation is usually visible. Enchondromas are frequently present as clusters. This leads to the metaphyseal widening. When they are localized at the bone border, they produce a typical notch-like image. A delay in bone age (average 0.6 +/- 1.3 years), has been reported in children with Ollier disease (7).
Enchondromas are usually localized in the metaphysis of long bones and in the small bones of the feet and hands. Initially, they are localized close to the growth plate cartilage. Later they migrate progressively towards the diaphysis. There may be irregularities in the epiphyseal region next to an affected metaphysis (1,6).
There is irregular distribution of the lesions. They can be localized to one limb, or limited to one half of the body. Though the lesions are limited largely to one side of the body, one or two enchondromas are frequently present on the other side, especially in the hand bones. When the lesions are distributed over the entire body, one side is usually more affected. The lesions in the hand almost never affect the metacarpal bones and phalanges.
Enchondromas can produce severe growth abnormalities. The growth abnormalities can be more severe than those observed in multiple exostosis. The affected diaphysis is short and massively enlarged. There may be bending close to the metaphysis. Ulnar shortening is usually more than shortening of the radius. The fingers are often of irregular sizes. Evidence of pathological fractures may be present.
Evidence of malignant transformation should be sought as it is a major complication of enchondromatosis. Signs of malignant transformation include extension of the tumor into soft tissues, cortical erosion, and irregularity or indistinctness of the surface of the tumour. Enchondromas are well circumscribed, chondrosarcomas on the other hand show poor demarcation. In differentiating enchondromas from chondrosarcomas the pattern of mineralization is also important. Enchondromas tend to show a uniform pattern of mineralization.
Histopathology
There are multiple oval-shaped or round cartilaginous nodules in osseous portions of bone on macroscopic examination of enchondromas (1,2). The nodules are limited at their periphery by lamellar or woven bone and are separated from each other by intertrabecular marrow spaces. The cartilaginous tumor matrix is usually solid. Myxoid changes, which manifest as frayings of the matrix are present. The presence of a striking heterogeneity and diversity in the degree of cellularity and chondrocyte phenotype characterizes enchondromas. This heterogeneity depends on factors such as localization and the patient's age. Due to this important cellular heterogeneity, the distinction between benign enchondromas and malignant chondrosarcomas is difficult. The histological criteria for malignancy that are used for conventional chondrosarcoma cannot be used in Ollier disease. This is because of the increased cellularity. Therefore the distinction between enchondroma and grade I chondrosarcoma in enchondromatosis is extremely difficult or even impossible. The diagnosis relies on a combination of radiographical, clinical, and histological criteria.
Etiology and pathogenesis
Endochondral bone ossification is a highly regulated process. It requires the progression of undifferentiated mesenchymal cells into hypertrophic chondrocytes and the subsequent replacement of a cartilaginous matrix by mineralized bone (8,9). Enchondromas develop in the metaphysis of long bones close to the growth plate. It has been proposed that they result from abnormalities in signaling pathways controlling the proliferation and differentiation of chondrocytes. This results in the development of intraosseous cartilaginous foci.
Genetics
Maffucci syndrome and Ollier disease are usually non-familial disorders (1-3). Both disorders tend to occur spontaneously and are not inherited. In Ollier disease, there is irregular distribution of the lesions. This strongly suggests that it is a disorder of endochondral bone formation that occurs due to a post-zygotic somatic mutation that results in mosaism. There have been two instances, where enchondromatosis has been observed in the sons of fathers who presented with mild skeletal dysplasia but without evidence of enchondromas (5,10). In one of these cases, there was a heterozygous mutation (R150C) in the PTH/PTHrP receptor (PTHR1 gene) that was inherited from the father (10).
Indian Hedgehog (IHH) and parathyroid hormone-related protein (PTHrP) act on their respective receptors PTHR1 and PTCH1 to exert a tightly coupled signaling relay, which is critical for the regulation of endochondral ossification. A study by Hopyan et al (10) showed that a mutant PTHR1 (R150C) was found to be expressed in enchondromas from two of six unrelated patients with enchondromatosis. The mutation was found on one parental allele in a patient and his father, who presented with mild skeletal dysplasia without enchondromatosis. However, in another study, neither the R150C mutation (26 tumors) nor any other mutation in the PTHR1 gene (11 patients) could be identified. This suggests that heterogeneity of the molecular defect(s) leads to enchondromatosis [11].
The mutant PTHR1 (R150C) seems to constitutively activate the PTHrP-dependent pathway. This decreases chondrocyte differentiation, thereby leading to the formation of enchondromas (10). Transgenic mice expressing the mutant PTHR1 under the control of the collagen type II promoter develop tumors that are similar to those observed in human enchondromatosis. Additional transgenic mice were generated that overexpress the Hedgehog (Hh) transcriptional regulator Gli2 because regulation of Ihh by PTHrP was found to be lost in these enchondromas. The mice developed ectopic cartilaginous islands similar to those observed in the mice expressing the mutant PTHR1. The Ihh signaling pathway plays a crucial role in the formation of enchondromatosis.
Cytogenetics and molecular genetics
There are few cytogenetic reports of benign enchondromas. There are no tumor-specific chromosomes or chromosomal regions associated with enchondromas, or chondrosarcomas (12-15).
Not much is known about the molecular mechanisms involved in the malignant transformation from enchondromas to chondrosarcomas. Expression of PTHR1, the PTHrP, and their downstream partner Bcl2 could be correlated with the grade of malignancy in chondrosarcoma (16-19).
Diagnosis
The diagnosis of Ollier disease is based on clinical and radiological evaluations. Histological analysis has a limited role. It is used if malignancy is suspected. Other investigations, such as ultrasound, scintigraphy, and magnetic resonance imaging (MRI) are not useful for establishing the diagnosis. They are useful for the evaluation and surveillance of lesions that become symptomatic i.e. cause pain or increase in size.
Differential diagnosis
Ollier disease has to be differentiated from HME (1-3). HME is an autosomal dominant disorder that is characterized by multiple bone tumors capped by cartilage which occur mostly in the metaphyses of long bones. Clinical and radiological criteria are used to establish the diagnosis of either disease. The most important criterion to distinguish enchondromas from osteochondromas as seen in HME is the localization of bone lesions. The osteochondromas are located at the bone surface and enchondromas are located in the center of bones. This allows radiographic distinction.
Other rare forms of chondromatosis, which include metachondromatosis, spondyloenchondroplasia, and genochondromatosis type I and II, have been described and defined well (1).
Treatment
There is no nonsurgical treatment for Ollier disease. Surgery is indicated when complications such as pathological fractures, growth defect, or malignant transformation occurs.
Prognosis
The prognosis of Ollier disease is usually difficult to assess (1). Patients with numerous lesions may have a better prognosis than patients with localized cartilaginous changes. These cartilaginous lesions can induce major shortening of a lower extremity and produce limb asymmetry, especially if already present in very young children. Early development of enchondromas in phalanges can lead to major finger deformities. Forms with an early onset are more severe. Neural compressions are less often observed than in HME. Enchondromas in Ollier disease can undergo malignant transformation into chondrosarcomas. This usually occurs in young adults. The reported incidence of malignant transformation is variable. It is estimated to occur in 5–50% of the cases (3,20-22). It is higher in Maffucci's syndrome. The prognosis is more severe than that in Ollier disease (1,2). Association of Ollier disease with other tumors has also been reported (1,23-25).
Conclusion
Enchondromas are common intraosseous, usually benign cartilaginous tumors. Its presentation is highly variable. It can range from an incidental finding to pathological fractures to limb length discrepancies. This variability can produce difficulty in making a diagnosis. The management is complicated and must be determined based on several factors. The primary goals of surgery in this disease are the correction of deformity and prevention of malignancy. The surgical treatment involves curettage, bone grafting, and in the more severe cases limb-lengthening or amputation.
References
Maroteaux P, Le Merrer M. Les maladies osseuses de l'enfant. Paris: Médecine-Sciences, Flammarion; 2002.
Unni KK. Cartilaginous lesions of bone. J Orthop Sci. 2001;6:457–472. doi: 10.1007/s007760170015.
Whyte M. Acquired Disorders of Cartilage and Bone. Washington DC: American Society for Bone and Mineral Research; 2003.
Fletcher CDM, Unni K, Mertens F, (Ed) World Health Organization Classification of Tumors Pathology and genetics Tumors of Soft Tissue and Bone. Lyon: IARC Press; 2002. p. 427.
Halal F, Azouz EM. Generalized enchondromatosis in a boy with only platyspondyly in the father. Am J Med Genet. 1991;38:588–592. doi: 10.1002/ajmg.1320380418.
Gabos PG, Bowen JR. Epiphyseal-metaphyseal enchondromatosis. A new clinical entity. J Bone Joint Surg Am. 1998;80:782–792.
Loder RT, Sundberg S, Gabriel K, Mehbod A, Meyer C. Determination of bone age in children with cartilaginous dysplasia (multiple hereditary osteochondromatosis and Ollier's enchondromatosis) J Pediatr Orthop. 2004;24:102–108.
Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657.
Schipani E, Provot S. PTHrP, PTH, and the PTH/PTHrP receptor in endochondral bone development. Birth Defects Res Part C Embryo Today. 2003;69:352–362. doi: 10.1002/bdrc.10028.
Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu C, Cole WG, Bell RS, Juppner H, Andrulis IL, Wunder JS, Alman BA. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat Genet. 2002;30:306–310. doi: 10.1038/ng844.
Rozeman LB, Sangiorgi L, Briaire-de Bruijn IH, Mainil-Varlet P, Bertoni F, Cleton-Jansen AM, Hogendoorn PC, Bovee JV. Enchondromatosis (Ollier disease, Maffucci syndrome) is not caused by the PTHR1 mutation p.R150C. Hum Mutat. 2004;24:466–473. doi: 10.1002/humu.20095.
Bovee JV, Cleton-Jansen AM, Kuipers-Dijkshoorn NJ, van den Broek LJ, Taminiau AH, Cornelisse CJ, Hogendoorn PC. Loss of heterozygosity and DNA ploidy point to a diverging genetic mechanism in the origin of peripheral and central chondrosarcoma. Genes Chromosomes Cancer. 1999;26:237–246. doi: 10.1002/(SICI)1098-2264(199911)26:3<237::AID-GCC8>3.0.CO;2-L.
Bovee JV, van Roggen JF, Cleton-Jansen AM, Taminiau AH, van der Woude HJ, Hogendoorn PC. Malignant progression in multiple enchondromatosis (Ollier's disease): an autopsy-based molecular genetic study. Hum Patho. 2000;31:1299–1303. doi: 10.1053/hupa.2000.19308.
Sandberg AA. Genetics of chondrosarcoma and related tumors. Curr Opin Oncol. 2004;16:342–354. doi: 10.1097/01.cco.0000129678.72521.e5.
Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: chondrosarcoma and other cartilaginous neoplasms. Cancer Genet Cytogenet. 2003;143:1–31. doi: 10.1016/S0165-4608(03)00002-5.
Amling M, Posl M, Hentz MW, Priemel M, Delling G. PTHrP and Bcl-2: essential regulatory molecules in chondrocyte differentiation and chondrogenic tumors. Verh Dtsch Ges Pathol. 1998;82:160–169.
Bovee JV, van den Broek LJ, Cleton-Jansen AM, Hogendoorn PC. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab Invest. 2000;80:1925–1934.
Kunisada T, Moseley JM, Slavin JL, Martin TJ, Choong PF. Co-expression of parathyroid hormone-related protein (PTHrP) and PTH/PTHrP receptor in cartilaginous tumours: a marker for malignancy? Pathology. 2002;34:133–137. doi: 10.1080/003130201201117936.
Pateder DB, Gish MW, O'Keefe RJ, Hicks DG, Teot LA, Rosier RN. Parathyroid hormone-related Peptide expression in cartilaginous tumors. Clin Orthop. 2002:198–204.
Rozeman LB, Hogendoorn PC, Bovee JV. Diagnosis and prognosis of chondrosarcoma of bone. Expert Rev Mol Diagn. 2002;2:461–472. doi: 10.1586/14737159.2.5.461.
Schaison F, Anract P, Coste F, De Pinieux G, Forest M, Tomeno B. Chondrosarcoma secondary to multiple cartilage diseases. Study of 29 clinical cases and review of the literature. Rev Chir Orthop Reparatrice Appar Mot. 1999;85:834–845.
Schwartz HS, Zimmerman NB, Simon MA, Wroble RR, Millar EA, Bonfiglio M. The malignant potential of enchondromatosis. J Bone Joint Surg Am. 1987;69:269–274.
Mahafza WS. Multiple enchondromatosis Ollier's disease with two primary brain tumors. Saudi Med J. 2004;25:1261–1263.
Tamimi HK, Bolen JW. Enchondromatosis (Ollier's disease) and ovarian juvenile granulosa cell tumor. Cancer. 1984;53:1605–1608. doi: 10.1002/1097-0142(19840401)53:7<1605::AID-CNCR2820530731>3.0.CO;2-N.
Vaz RM, Turner C. Ollier disease (enchondromatosis) associated with ovarian juvenile granulosa cell tumor and precocious pseudopuberty. J Pediatr. 1986;108:945–947. doi: 10.1016/S0022-3476(86)80936-2.
No comments:
Post a Comment